Gene Technologies
Genome sequencing
The genome is all of the genes present in an organism. The human genome, as well as the genomes of thousands of other living organisms, has been sequenced. Sequencing is only accurate for short sections of DNA, so DNA first needs to be made into shorter fragments. These individual fragments are sequenced and then the genome can be put together.
Once we know the sequence of DNA bases that make up a particular gene, we can determine the amino acid sequence and structure of the protein that it codes for. This is useful for medical research – for example, it allows scientists to determine the shape of antigens on disease-causing viruses based on its DNA sequence. Knowing antigen structure helps to develop an effective vaccine.
From the genome, we can determine the proteome (all of the proteins present in an organism). This is easier for simpler organisms such as bacteria, which have less non-coding DNA. Humans and other more complicated organisms have more non-coding DNA e.g. introns, promoters) so scientists first have to sort through the genome and work out which bits code for proteins. Only once they have identified the coding DNA can they determine the genome.
Over the past few decades, sequencing techniques have improved, speeding up the process and reducing the cost.
Making DNA fragments
If you want to sequence the genome, you first have to chop it up into smaller pieces because the sequencing method won’t work on such a big section of DNA in one go. Scientists also need to produce DNA fragments if they want to take a useful gene from one species (e.g. for heat-tolerance) and place it in another species. This is known as genetic engineering and the modified organism is referred to as transgenic.
Methods of making DNA fragments:
Restriction enzymes
Restriction enzymes are enzymes that cut double-stranded DNA at specific recognition sequences.
The recognition sites are palindromic – they read the same backwards and forwards.
When the restriction enzyme detects its recognition sequence, it cuts DNA in one of two ways – either straight down the middle, creating ‘blunt ends’ or in a zig-zag fashion, creating ‘sticky ends’. Sticky ends are overhangs of single-stranded DNA at the ends of the fragment. Different restriction enzymes have different recognition sequences. They cut the double-stranded DNA by hydrolysing the phosphodiester backbone.
Scientists will first analyse the DNA to determine which recognition sites are present either side of the gene of interest. They will then use the corresponding enzymes to extract the gene from the longer section of DNA.

Reverse transcriptase
It’s often easier to extract mRNA from cells instead of DNA, because there are more copies.
mRNA is reverse-transcribed into complementary DNA (cDNA) using an enzyme called reverse transcriptase.
The extracted mRNA is mixed with DNA nucleotides and reverse transcriptase. The enzyme uses the mRNA as a template to synthesise the complementary DNA.
A ‘gene machine’
DNA fragments can be synthesised from scratch by fixing a nucleotide onto a solid support e.g. a bead.
Nucleotides are individually added in the correct order.
Protecting groups are added to prevent branching of the nucleotide chain.
Oligonucleotides, around 20 base pairs long, are synthesised. The protecting groups are removed and multiple oligonucleotides can be joined to make a longer DNA fragment.
Making a transgenic organism
To make a transgenic organism, you now need to deliver the DNA fragment (containing the gene of interest) into the organism using a vector. Common vectors include plasmids and bacteriophages.
The plasmid is cut using the same restriction enzymes that we used to extract the gene – this creates complementary sticky ends.
The fragment is inserted into the plasmid using DNA ligase – an enzyme which anneals the complementary sticky ends in a process called ligation.
The modified plasmid is referred to as recombinant DNA as it contains genetic material from two different sources.
The recombinant plasmid can now be inserted into a host cell:
- The plasmid is mixed with bacterial cells and encouraged to take up the vector using heat shock. The mixture is placed on ice then heated to 42oC from a few minutes. This increases the permeability of the cell wall, allowing it to absorb the plasmid.
- If you are using a bacteriophage, all you need to do is to mix it with the bacterial cells. The phage will naturally infect the cells, injecting its DNA into them.
- Any cells that take up the recombinant DNA are said to be transformed.
- This process is inefficient – so only a small proportion of the bacterial population will be transformed.
Identifying transformed cells relies on the fact that the plasmid you used contains a marker gene e.g. a gene for antibiotic resistance. This means that any cells that contain the gene of interest will also be resistant to the antibiotic.
Grow the bacteria on an agar plate containing the antibiotic.
Only transformed cells will survive.
Surviving colonies are selected and grown on a larger scale.
Other marker genes may also be used e.g. a gene that codes for a fluorescent protein. When the agar plate is placed under UV light, only the transformed bacteria will glow.
As well as a marker gene, the vector also needs to contain promoter and terminator regions for the organism synthesise a protein. Promoter regions allow RNA polymerase to bind and initiate transcription, while terminator regions tell it to stop transcribing.
Polymerase Chain Reaction (PCR)
PCR is a technique used to amplify fragments of DNA. This is important because the DNA sample collected at a crime scene doesn’t contain enough material for accurate analysis. Before PCR is carried out, you need to prepare a mixture containing the following:
DNA sample
Free DNA nucleotides
Primers (these are short pieces of DNA which bind to the beginning of the DNA fragment and initiate replication)
The enzyme DNA polymerase which has been extracted from thermophilic (heat-loving) bacteria.
PCR is then carried out in the following stages:
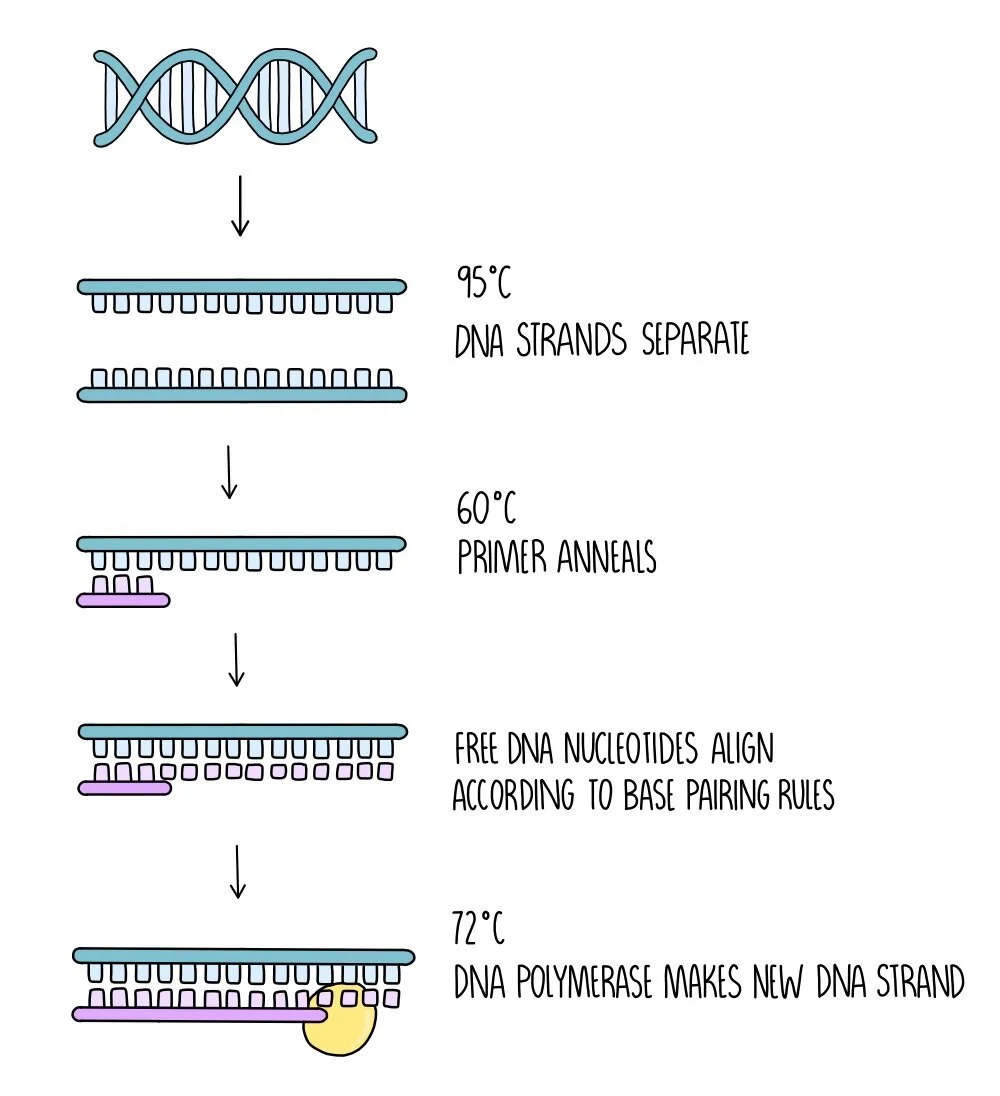
Separation of the DNA strands - the mixture is heated to 95oC which causes hydrogen bonds between the DNA strands to break.
Annealing of the primer - the mixture is then cooled to around 60oC which allows the primer to anneal to the DNA.
DNA synthesis - The temperature is increased to 72oC which is the optimum temperature for DNA polymerase. DNA polymerase forms a new DNA strand from catalysing the formation of phosphodiester bonds between the free DNA nucleotides which align along the DNA template strand by complementary base pairing rules.
Around 30-40 cycles of PCR are carried out, which generates millions of DNA fragments. Each PCR cycle doubles the amount of DNA, so huge numbers of DNA fragments can be quickly generated.
Making drugs using recombinant DNA technology
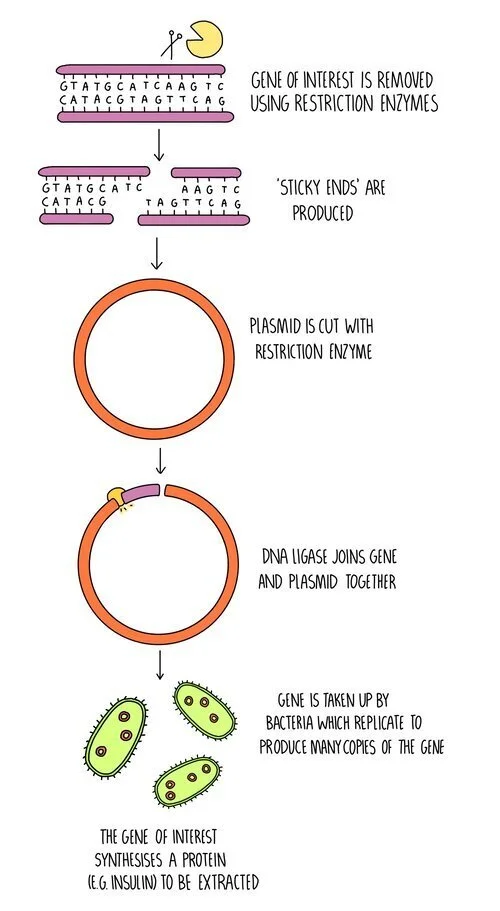
Drugs such as insulin are made using genetically-modified bacteria in the following process:
The insulin gene is removed from human DNA using restriction enzymes.
A plasmid is also cut with restriction enzymes.
DNA ligase joins the complementary sticky ends to form the recombinant DNA.
The recombinant plasmid is taken up by bacteria.
The transgenic bacteria are grown in large fermenters to produce large amounts of insulin, which can then be extracted.
Bacteria aren’t the only organisms which can be genetically modified to produce drugs - plants and animals can be used too. For genetically modifying plants, a GM bacterium is first made using the process outlined above. The bacterium then acts as a vector, infecting a plant cell and inserting its DNA into the genome of the plant cell. The plant cell grows into an adult plant and all of its cells will contain the drug-producing gene. The plant cells will synthesise the protein which can either be extracted or the drug can be delivered to the patient by eating the plant. Cholera vaccines have been made using this method.
Genetically-modified animals are produced by injecting the gene for the protein (which will act as the drug) into the nucleus of a fertilised animal egg cell. This is then implanted into an adult animal and as the animal develops, every cell will contain the drug-producing gene. The protein produced from the gene is normally purified from the milk of the animal. This method has been used in goats to produce the drug antithrombin for treating people with defective blood clotting.
Issues with using GMOs in medicine and food production
GMOs are not just used for producing drugs, but also for improving agriculture. For example, genes can be added to crop plants to make them resistant to disease or more nutritious. However, the use of GMOs is controversial and there are some arguments for and against their use in medicine and agriculture:
Arguments for the use of GMOs:
Genetic modification can increase crop yield and make food more nutritious - this means that farmers can make more money and people will be healthier.
Genes for pest-resistance can be introduced into crops. This means that farmers save money on pesticides and the environmental problems associated with pesticide use are reduced.
Human proteins produced using GMOs do not result in allergic reactions - for example, insulin as a treatment for diabetes used to be extracted from the blood of pigs and cows. Nowadays human insulin is produced using GM bacteria which doesn’t produce an allergic reaction in patients and is more efficient.
Vaccines produced using genetically-modified plants do not need to be kept cold to stay effective - this is an advantage when vaccines are needed in remote regions where refrigeration is not possible.
Enzymes can be produced using GMOs and are often used in industrial processes e.g. in the manufacture of washing detergents and the textile industry. The use of GMOs makes these processes cheaper.
The process is cheap because once one genetically modified organism is made, lots more can be made through breeding the original GM organism. This makes drugs cheaper.
Arguments against the use of GMOs:

Protesters in Japan march to show their opposition to GM foods. Credit: Alessandro Di Ciommo/Nurphoto (Getty Images)
GMOs are often patented and the seeds are expensive to buy - this puts farmers in developing countries at a disadvantage.
If a GM plant cross-pollinated with another species, it could introduce the genes into other plants with unintended consequences. For example, cross-breeding a herbicide-resistant crop plant with wild plant species could create ‘superweeds’ which are resistant to herbicides. These could out-compete other plants with potential effects on the whole food chain.
There are ethical issues against the use of GMOs - should humans manipulate animals for our own benefit?
Religious reasons - do humans have the right to ‘play God’ and create new life?
Some people worry out the long-term impact of using GMO - there may be unforeseen consequences which are impossible to predict.
Gene therapy
Gene therapy is a method used to treat genetic diseases. For recessive diseases, a functional copy of the gene is introduced into the patient’s cells using a vector (usually viruses or liposomes). Since the cells now contain a working copy of the gene, they will synthesise the functional protein which should reduce the symptoms of the disease. For dominant diseases, the mutated allele can be silenced by inserting a piece of DNA in the middle of the gene to stop it from being expressed.
There are two types of gene therapy:
Germ line – the allele is inserted into a gamete. This has the advantage that all of the cells will contain the functional allele. However, it will be passed onto offspring, making it controversial. It is currently illegal.
Somatic – the allele is inserted into the body cells that are affected by the disorder. It only changes a handful of cells and will need to be repeated when those cells eventually die. It is being used as a treatment in a limited capacity.
DNA probes
DNA probes are used to see whether a person contains a specific allele e.g. the mutated CFTR allele that causes cystic fibrosis in homozygous individuals. The probe itself is a short strand of DNA which has a complementary base sequence to the allele you’re interested in. If the allele is present, it will bind to the probe. The probe is attached to a fluorescent tag or a radioactive label, to allow detection.
Method:
Restriction enzymes digest the DNA sample into fragments which are separated by electrophoresis.
Separated fragments are transferred to a nylon membrane and incubated with the fluorescent DNA probe.
If the allele is present, the fluorescent probe will anneal (bind) to it.
The membrane is visualised under UV light – if the allele is present, it will appear as a fluorescent band. This tells us that the person who provided the DNA sample has the allele.
A similar technique is a DNA microarray, which allows us to analyse the presents of multiple genes at the same time:
A plate is used containing lots of wells – each well will have a different DNA probe attached.
To synthesise the DNA probes, you find need to know the sequence of the alleles you are interested in. PCR is then used to synthesise the complementary probes.
A sample of fluorescently-labelled DNA is washed over the array (note this time, the DNA sample is tagged, rather than the probe).
Any DNA that is complementary to the probes will anneal to the array.
The plate is washed to remove unbound DNA.
The plate is visualised under UV light – any DNA attached to a probe will appear as a fluorescent dot.
Screening for disease alleles has multiple uses:
To diagnose genetic diseases, such as cystic fibrosis.
To determine how a patient will respond to specific drugs – e.g. people with breast cancer respond differently to different drugs depending on the mutation – knowing which mutation is present helps doctors to choose the best course of treatment
To identify health risks – e.g. some people may have a mutation that makes them more likely to develop cardiovascular disease. This may encourage them implement lifestyle changes that prevent them from developing the disease. There is a risk that insurance companies and employers may use this kind of data to discriminate against people.
DNA Profiling
DNA profiling is a technique which can be used to analyse a sample of DNA (e.g. one found at a crime scene) and compare it to DNA samples taken from the suspects.
The DNA sample will be collected (this is usually blood, saliva or semen) and amplified using PCR.
The PCR products are separated using gel electrophoresis, which separates the DNA fragments according to length.
The gel is visualised using UV light and the banding patterns from the suspect’s DNA can be compared with that found at the crime scene.
The same technique can also be used to identify genetic relationships between people (as in paternity testing) or to determine evolutionary relationships between organisms.
Electrophoresis
So that the DNA can be visualised, we add a fluorescent molecule which binds to the DNA and makes it visible when exposed to UV light. A common fluorescent tag is ethidium bromide, which inserts itself between the DNA bases and gives off fluorescence under UV light.
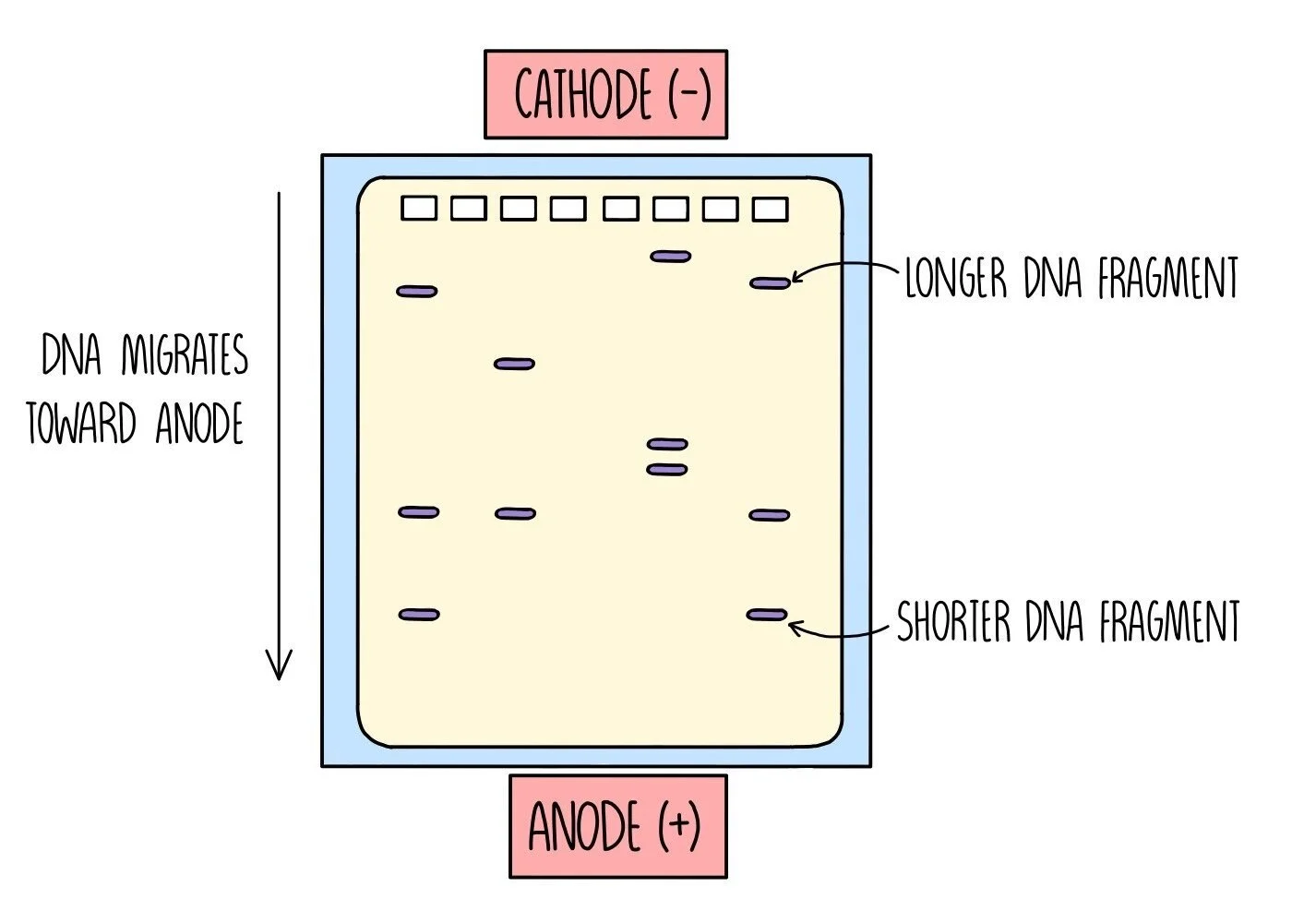
Once the DNA fragments have been amplified and stained with a fluorescent dye, they need to be separated. This is done using gel electrophoresis, which separates the DNA strands according to length. It works because DNA is negatively charged, which means it will move towards a positive charge when placed in an electric field. Shorter DNA fragments travel through the gel more quickly, which means they will travel a longer distance than the larger DNA fragments.
Gel electrophoresis is carried out in the following steps:
An agarose gel is prepared which contains a row of wells at the top of the gel. The gel is placed into a tank containing buffer solution which is able to conduct electricity.
The DNA sample is mixed with a loading dye - this turns the DNA mixture a dark colour and helps you see what you’re doing. A fixed volume of the DNA samples are pipetted into the wells.
An electrical current is passed through the gel and the DNA will begin to move towards the bottom of the gel (towards the anode).
Once the dye has reached the bottom, the electricity is turned off and the banding pattern is visualised under UV light.